class: center, middle, inverse, title-slide # scFeatureFilter: ## correlation-based feature filtering for single-cell RNA-seq ### <span class="citation">@G_Devailly</span> ### 2018/01/17 --- # Introduction - Single cell RNA-sequencing is increasingly popular. - scRNA-seq is noisier than bulk RNA-seq. - Filtering of noisy, lowly-expressed features.red[*] is common. .footnote[.red[*]Feature: gene or transcript] --- ## Introduction - Using spike-in RNA information - Arbitrary filtering: >.small[[...] on a filtered data set, where we retain only genes with an estimated TPM above 1 in more than 25% of the considered cells. (1)] >.small[Genes with less than 5 reads and expressed in less than 10 cells were removed. (2)] >.small[Here, low-abundance genes are defined as those with an average count below a filter threshold of 1 [count]. (3)] >.small[ Genes were filtered, keeping 15,633 out of 26,178 genes that were expressed in at least 5 out of 1,919 sequenced cells (RPKM ≥ 10) and for which cells with expression came from at least two different embryos. (4)] .tiny[.red[1] Soneson & Robinson, bioRxiv, 2017 .red[2] Stevant et al., bioRxiv, 2017 .red[3] Lun et al., F1000Research, 2016 .red[4] Petropulos et al., Cell, 2016] --- ## Introduction - No standard threshold for filtering. - Same threshold might not be of the *same stringency* in different datasets, notably across species.red[*]. .footnote[.red[*] See Mansoki et al., Comput Biol Chem., 2016] * Can we do better? --- class: inverse # scFeatureFilter - R package - Available on GitHub: [github.com/gdevailly/scFeatureFilte](https://github.com/gdevailly/scFeatureFilter) - Accepted in [Bioconductor](https://www.bioconductor.org/packages/devel/bioc/html/scFeatureFilter.html) .footnote[Need R ≥ 3.5 (*or edit DESCRIPTION to depends: R ≥ 3.4*)] - Might help to set a relevant expression threshold for feature filtering. ```r library(scFeatureFilter) ``` --- ## Example datasets: 32 scRNA-seq of human embryonic stem cells .small[(Yan et al., Nat Struct Mol Biol, 2013.)] ```r dim(scData_hESC) ## [1] 60468 33 ``` ```r scData_hESC ``` .small[ |gene | cell_1| cell_2| cell_3| cell_4| |:------------------|------:|------:|------:|------:| |ENSG00000000003.13 | 55.33| 35.98| 53.68| 31.95| |ENSG00000000005.5 | 0.00| 0.00| 0.13| 0.00| |ENSG00000000419.11 | 53.97| 55.47| 41.87| 110.75| |ENSG00000000457.12 | 0.92| 0.22| 0.65| 0.87| ] .small[Expression matrices can be either `data.frame`, `tibble`, `matrix` or `SingleCellExperiment`.] --- ## Mean-variance exploration: ```r calculate_cvs(scData_hESC, max_zeros = 0.75)[1:4, 1:5] ## # A tibble: 4 x 5 ## geneName mean sd cv ## <chr> <dbl> <dbl> <dbl> ## 1 ENSG00000000003.13 70.022328 73.332127 1.0472678 ## 2 ENSG00000000005.5 1.000291 2.527535 2.5267987 ## 3 ENSG00000000419.11 80.991847 71.534922 0.8832361 ## 4 ENSG00000000457.12 1.590995 1.804686 1.1343132 ## # ... with 1 more variables: GSM922224_hESCpassage0_Cell4_0 <dbl> ``` `max_zeros`: maximum proportion of `0` value for a feature to be kept --- ## Mean-variance exploration: ```r calculate_cvs(scData_hESC, max_zeros = 0.75) %>% plot_mean_variance(colourByBin = FALSE) + annotation_logticks(sides = "l") ``` <img src="devailly_scFeatureFilter_files/figure-html/meanVar1-1.png" width="56%" style="display: block; margin: auto;" /> --- ## Binning of the genes: ```r scData_hESC %>% calculate_cvs %>% define_top_genes(window_size = 100) %>% bin_scdata(window_size = 1000) %>% plot_mean_variance() + annotation_logticks(sides = "l") ``` <img src="devailly_scFeatureFilter_files/figure-html/meanVar2-1.png" width="56%" style="display: block; margin: auto;" /> --- ## Assumptions .pull-left[ <img src="devailly_scFeatureFilter_files/figure-html/meanVar3-1.png" width="100%" style="display: block; margin: auto;" /> ] .pull-right[ - high expression = less technical variation - biological variation = transcription module - correlation of genes belonging to the same transcription module ] --- ## Correlation of the data: - **A reference set of genes**: the top bin - Three control sets of genes: shuffling of the expression values of the reference set Each gene in each bin is correlated against each gene in the **reference set**, and each gene in the 3 **control sets**: ```r corDistrib <- scData_hESC %>% calculate_cvs %>% define_top_genes(window_size = 100) %>% bin_scdata(window_size = 1000) %>% correlate_windows(n_random = 3) ## Mean expression of last top gene: 2114.40221875 ## Number of windows: 18 ``` --- ## Correlation of the data ```r corDens <- correlations_to_densities(corDistrib) plot_correlations_distributions(corDens, facet_ncol = 6) + scale_x_continuous(breaks = c(0, 0.5, 1), labels = c("0", "0.5", "1")) ``` <img src="devailly_scFeatureFilter_files/figure-html/plotCor-1.png" width="80%" style="display: block; margin: auto;" /> --- ## Correlation of the data ```r metrics <- get_mean_median(corDistrib) plot_correlations_distributions(corDens, metrics = metrics, facet_ncol = 6) + scale_x_continuous(breaks = c(0, 0.5, 1), labels = c("0", "0.5", "1")) ``` <img src="devailly_scFeatureFilter_files/figure-html/plotCor2-1.png" width="80%" style="display: block; margin: auto;" /> --- ## Threshold decision ```r plot_metric(metrics, show_ctrl = FALSE, show_threshold = FALSE) ``` <img src="devailly_scFeatureFilter_files/figure-html/threshold1-1.png" width="76%" style="display: block; margin: auto;" /> --- ## Threshold decision ```r plot_metric(metrics, show_ctrl = TRUE, show_threshold = FALSE) ``` <img src="devailly_scFeatureFilter_files/figure-html/threshold2-1.png" width="76%" style="display: block; margin: auto;" /> --- ## Threshold decision ```r plot_metric(metrics, show_ctrl = TRUE, show_threshold = TRUE, threshold = 2) ``` <img src="devailly_scFeatureFilter_files/figure-html/threshold3-1.png" width="76%" style="display: block; margin: auto;" /> --- ## Geting back the filtered expression matrix: ```r binned_data <- scData_hESC %>% calculate_cvs %>% define_top_genes(window_size = 100) %>% bin_scdata(window_size = 1000) ``` ```r determine_bin_cutoff(metrics, threshold = 2) ## [1] 9 filtered_data <- filter_expression_table( binned_data, bin_cutoff = determine_bin_cutoff(metrics) ) nrow(scData_hESC) ## [1] 60468 nrow(binned_data) ## [1] 17929 nrow(filtered_data) ## [1] 7442 ``` --- # A shortcut: ```r filtered_data <- sc_feature_filter(scData_hESC) ## Mean expression of last top gene: 2114.40221875 ## Number of windows: 18 dim(scData_hESC) ## [1] 60468 33 dim(filtered_data) ## [1] 7442 33 ``` --- class: inverse # Testing `scFeatureFilter` --- class: center ## Testing `scFeatureFilter` on more datasets: .medium[16 datasets from [ConquerDB](http://imlspenticton.uzh.ch:3838/conquer/) (human and mouse)] 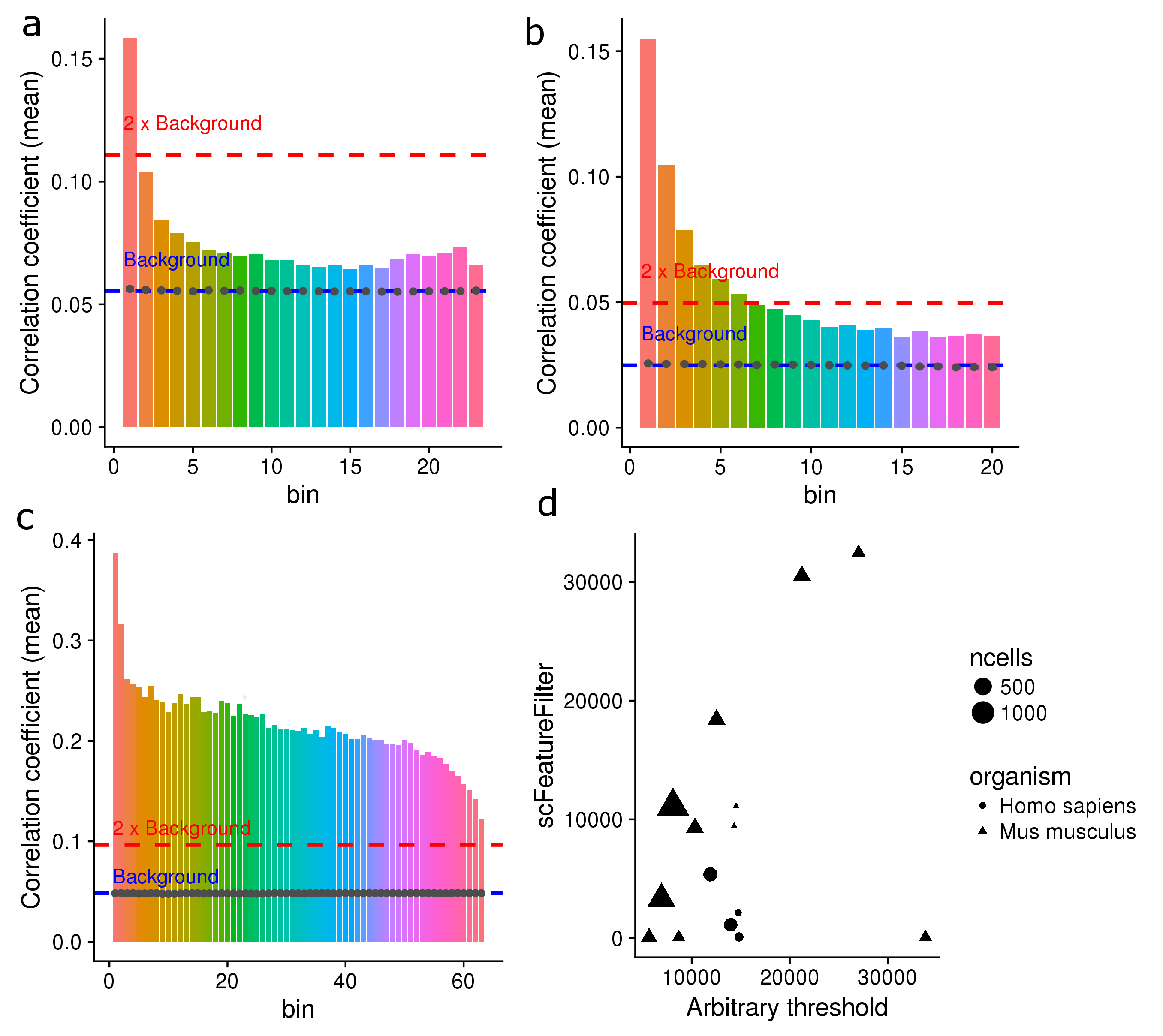 --- class: center ## `scFeatureFilter` on bulk RNA-seq: .medium[48 repplicated bulk RNA-seq (yeast)] .tiny[(Gierlinski et al., Bioinformatics, 2015)] 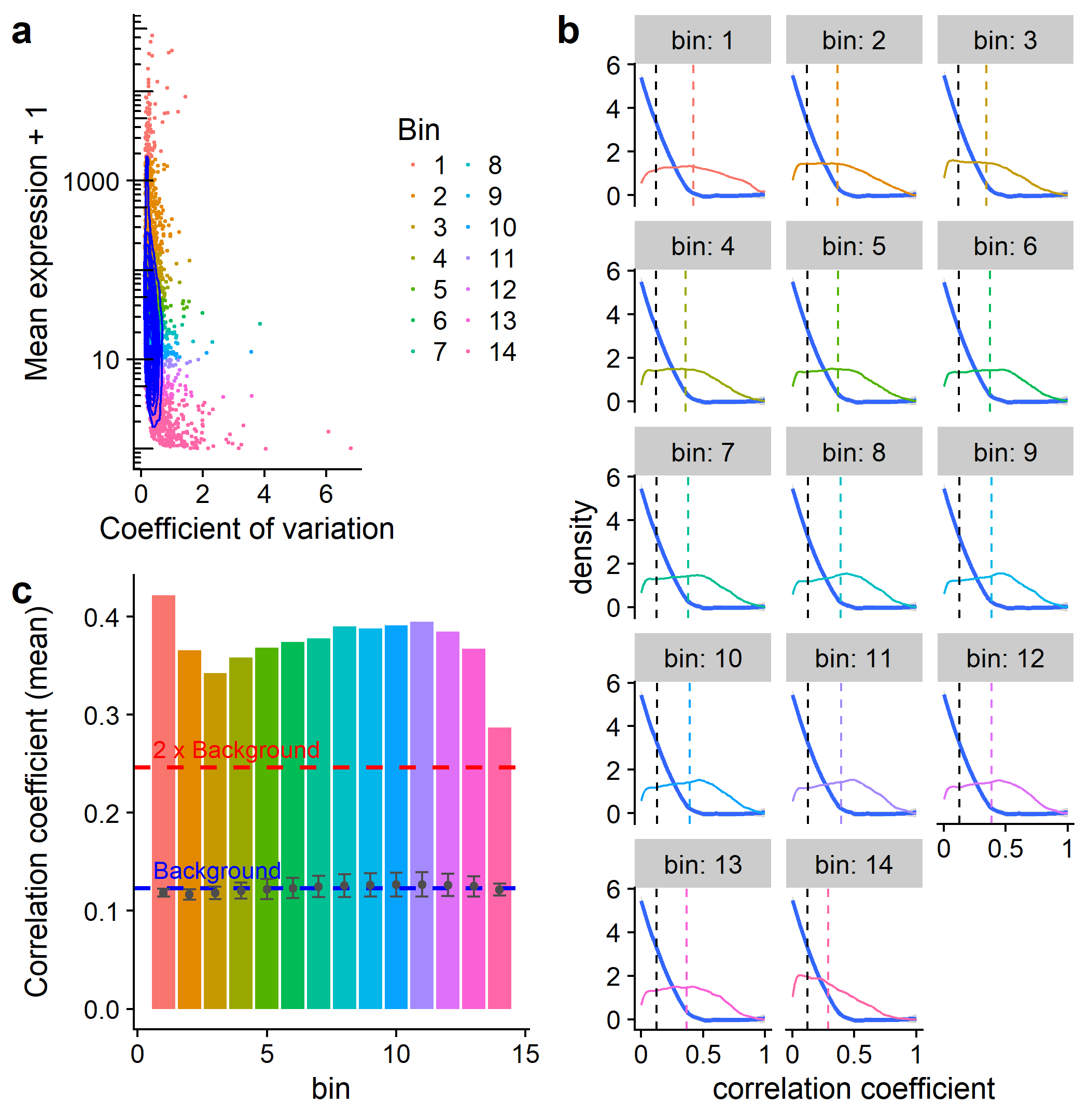 --- class: inverse # Limits of `scFeatureFilter` --- ## Lots of parameters? .medium[ |parameter |description |default| |:-------------------|:-------------------------------------------------|------:| |`max_zeros` | maximum proportion of 0 for a feature to be kept | 0.75| |`top_window_size` | size of the reference set | 100| |`other_window_size` | size of the other bins | 1000| |`threshold` | stringency of the selection | 2| ] --- ## Lots of parameters? The method is robust to `other_window_size`: <img src="devailly_scFeatureFilter_files/figure-html/winSize-1.png" width="100%" style="display: block; margin: auto;" /> --- ## Robust to `max_zeros`? high proportion of 0s ~= low expression - mostly not in the reference set - more abundant in the low expression bins - less abundant in the high expression bins ## `threshold` is a feature `threshold`: More or less stringency depending of the use cases and user preference. --- ## `top_window_size` `top_window_size` can have *massive* impact: - if **too big**: Risk of selecting everything. - if **too small**: Might not capture enough biological variation. ~100 seems to be a sweet spot on mouse and human data. --- ## `top_window_size` Average auto-correlation of the top window depending on its size: ```r plot_top_window_autocor(calculate_cvs(scData_hESC)) ``` <img src="devailly_scFeatureFilter_files/figure-html/topWinSize-1.png" width="55%" style="display: block; margin: auto;" /> --- # Other limits? - Tested on a dataset with 1378 cells. Scalable until when? - Not designed nor tested for 10x genomics scRNA-seq. --- class: inverse # Conclusion: `scFeatureFilter`: an R package for less arbitrary threshold selection We are looking for feedback: - Usefull? - Overkilled? - Broken assumptions? - Better existing methods? --- class: inverse .pull-left[ # Thanks Anagha Joshi Angeles Arzalluz-Luque Anna Mantsoki ] .pull-right[  ]